pycombat_norm: batch effect corrections for microarray data
ComBat [Johnson2007] is one of the most widely used tool for batch
effect correction for microarray transcriptomic data.
pycombat_norm() is a Python implementation of ComBat. Strictly following
the same mathematical framework, pycombat_norm() has results similar to
those of ComBat in terms of batch effects correction. Additionally,
pycombat_norm() is as fast, if not faster, than the original
implementation in R.
Minimal usage example
This minimal usage example illustrates how to use pycombat_norm in a default setting, and shows some results on ovarian cancer data. The data we use is freely available on NCBI Gene Expression Omnibus, namely:
GSE18520
GSE66957
GSE69428
The corresponding expression files are stored on InMoose repository in the data subfolder.
# import libraries
from inmoose.pycombat import pycombat_norm
import pandas as pd
import matplotlib.pyplot as plt
# prepare data
# the datasets are dataframes where:
# the indices correspond to the gene names
# the column names correspond to the sample names
# Any number (>=2) of datasets can be treated at once
dataset_1 = pd.read_pickle("data/GSE18520.pickle")
dataset_2 = pd.read_pickle("data/GSE66957.pickle")
dataset_3 = pd.read_pickle("data/GSE69428.pickle")
# merge all three datasets into a single one, keeping only common genes
df_expression = pd.concat([dataset_1,dataset_2,dataset_3],join="inner",axis=1)
# plot raw data
plt.boxplot(df_expression)
plt.show()
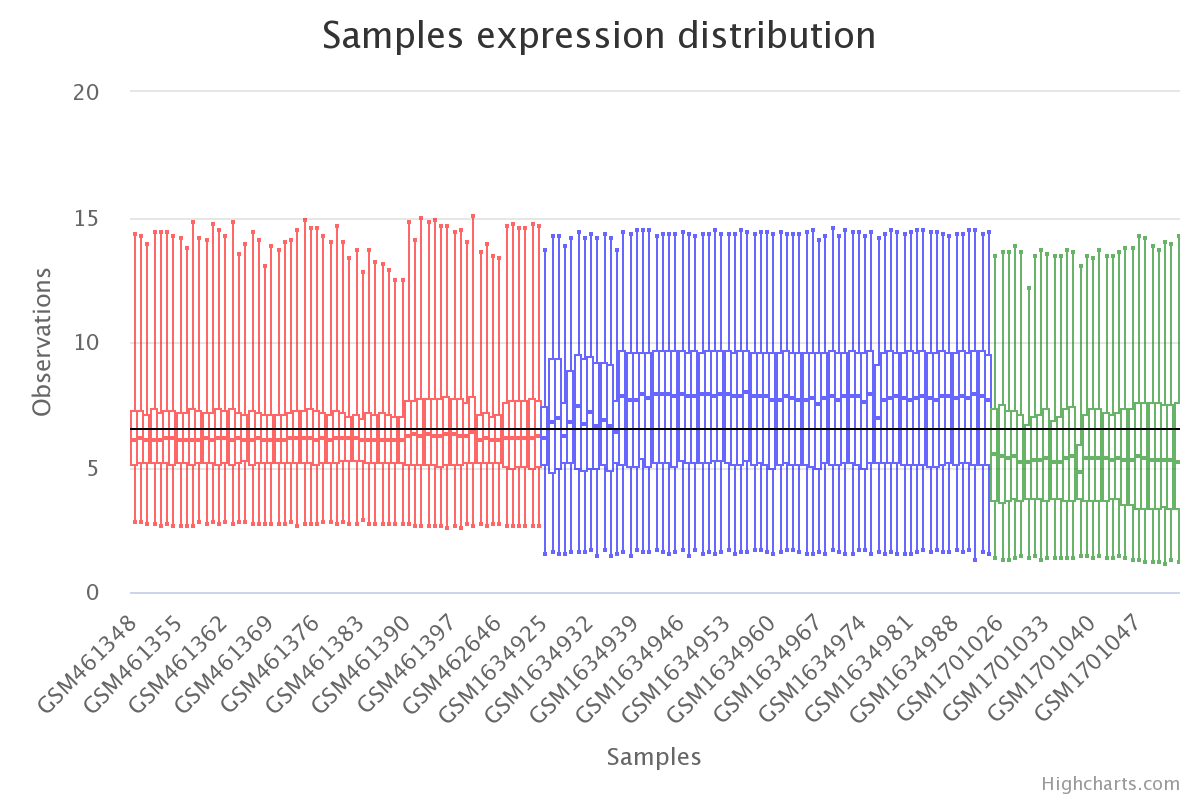
Gene expression by sample in the raw data (colored by dataset).
# generate the list of batches
datasets = [dataset_1, dataset_2, dataset_3]
batch = [j for j,ds in enumerate(datasets) for _ in range(len(ds.columns))]
# run pycombat_norm
df_corrected = pycombat_norm(df_expression, batch)
# visualize results
plt.boxplot(df_corrected)
plt.show()
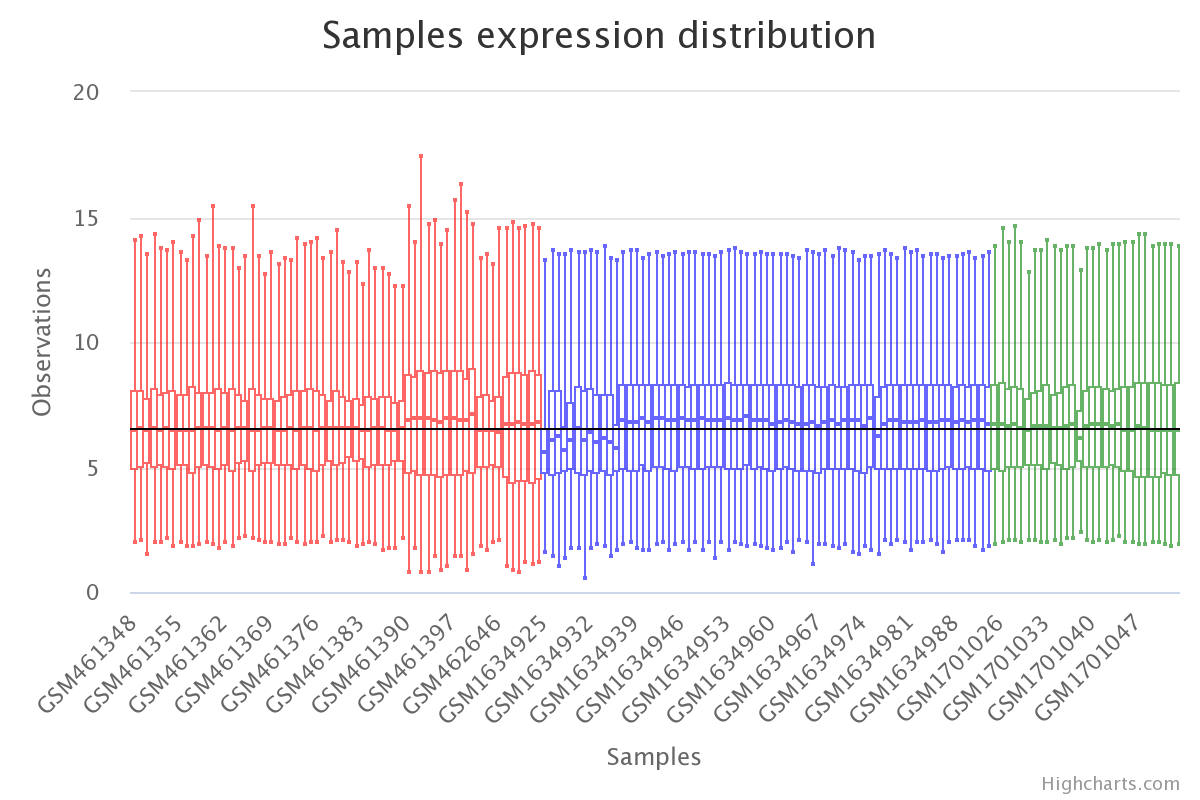
Gene expression by sample in the batch effect-corrected data (colored by dataset).
Biological Insight
The data we used for the example above contain tumor samples and healthy samples. A simple PCA on the raw expression data shows that, instead of clustering by sample type, data cluster by dataset.
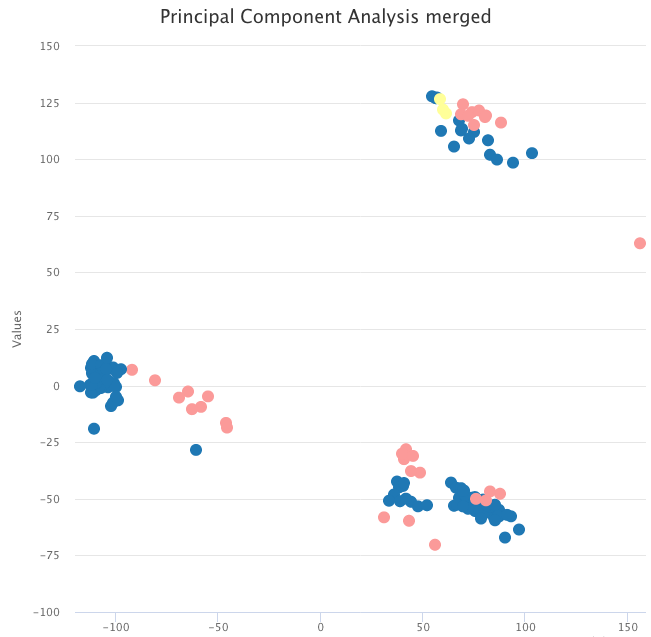
PCA on the raw expression data, colored by tumor sample (blue and yellow) and healthy sample (pink).
However, after correcting batch effects with pycombat_norm, the same PCA shows two clusters, corresponding respectively to tumor and healthy samples.

PCA on the batch effect-corrected expression data, colored by tumor sample (blue and yellow) and healthy sample (pink).
Code documentation
- inmoose.pycombat.pycombat_norm(counts, batch, covar_mod=None, par_prior=True, prior_plots=False, mean_only=False, ref_batch=None, precision=None, na_cov_action='raise', **kwargs)
Corrects batch effect in microarray expression data. Takes an gene expression file and a list of known batches corresponding to each sample.
- Parameters:
counts (np.ndarray or pd.DataFrame or ad.AnnData) – expression matrix. It contains the information about the gene expression (rows) for each sample (columns).
batch (list or str) – batch indices. Must have as many elements as the number of columns in the expression matrix. If
countsis an AnnData or a DataFrame,batchcan be the name of the column containing the batch data.covar_mod (list or matrix, optional) – model matrix (dataframe, list or numpy array) for one or multiple covariates to include in linear model (signal from these variables are kept in data after adjustment). Covariates have to be categorial, they can not be continuous values (default: None).
par_prior (bool, optional) – False for non-parametric estimation of batch effects (default: True).
prior_plots (bool, optional) – True if requires to plot the priors (default: False). – Not implemented yet!
mean_only (bool, optional) – True iff just adjusting the means and not individual batch effects (default: False).
ref_batch – batch id of the batch to use as reference (default: None)
optional – batch id of the batch to use as reference (default: None)
precision (float, optional) – level of precision for precision computing (default: None).
na_cov_action (str) –
Option to choose the way to handle missing covariates
"raise"raise an error if missing covariates and stop the code"remove"remove samples with missing covariates and raise a warning"fill"handle missing covariates, by creating a distinct covariate per batch
(default:
"raise")
- Returns:
the input expression matrix adjusted for batch effects. same type as the input data
- Return type:
matrix